















Here is an interesting quote I came across today. It is meant to help you solve problems:
"You write down the problem. You think very hard. Then you write down the answer. "
~Richard Feynman
OK, I am going to try it right now with a problem I have. I am hungry. I missed breakfast and have not had lunch yet (it's 1.15). But the isopropanol is evaporating sooooo slowly, we need to do other things once it finally goes but it's taking it's own sweet time.
Problem: I am hungry
Thinking: need food, really need food, but it's so cold outside and too far to walk and I can't be bothered and I need more money food, need food.
Answer (written after a minimum of three minutes thinking time): I will fill myself up with water and not be hungry.
...
Sort of worked. Try again:
Problem: I have a 3000 word essay to write before January 20th
Thinking: I've sort of started it, I know some stuff, I have a week at home where I can sort of work except I won't have the Internet (crap) I have the weekend except I should probably be spending that with a Certain Someone as it is my last week with them before the holidays. why is this useful anyway! Lab Rat's don't need to be able to write essays. at least I'll have 20 days when I get back, crap how am I supposed to do anything in 20 days. I can't think I'm too hungry!!
Answer: ...
Screw you Richard Feynman.
:(
Field of Science
-
-
Change of address1 year ago in Variety of Life
-
Change of address1 year ago in Catalogue of Organisms
-
-
Earth Day: Pogo and our responsibility1 year ago in Doc Madhattan

And why have you used 84 plates?
The thing about scientific protocols is that they are meant to be exact and precise. Every step must be explained concisely and there should be a reason for all the methodology.
For example, for every protocol you use, you should be able to answer random questions about why you did what you did at each step. Why was the bacteria incubated for four hours? Why was the temperature kept at 50 degrees? Why was the product stored on ice?
The answers to these questions should be sensible and scientific. Temperatures, amounts and incubation's are used to optimise reactions. Every step should be planned to get the best possible result in the most efficient way.
At the moment, we're growing phages on agar plates. At one stage of the protocol, we use exactly 84 plates to grow them. Two of the plates are controls and four are for dilutions but the fact remains that every time we use exactly 84 plates, no matter what we're doing.
The answer (as can probably be guessed) is not scientific in the least. Science is not a cold and clinical organised space, no matter how much scientists want it to be. It is a wild and crazy world full of human error and things going random and even more human error. The problems are not just scientific, they are also spacial and temporal; incubators are only so big, parts of the lab are only open during certain hours.
And the big jars we use for incubating will only physically fit 42 plates. We've tried to stuff more in but the lids won't shut. And while we have three of them in total the incubator is quite small and only fits two at a time.
42 x 2 = 84
Which wouldn't seem so bad if it weren't for the dilutions and the controls, because once you've put them in there's only room for 39 plates of actual phage sample per jar. 42 is at least a nice round number, but a protocol always looks a bit odd if it starts with the phrase '39 plates were taken...' It begs the question, 'why 39?' and the answer is, scientifically, faintly embarrassing.
Science would be a lot more precise if the world stopped getting in the way.
But far less fun =D
For example, for every protocol you use, you should be able to answer random questions about why you did what you did at each step. Why was the bacteria incubated for four hours? Why was the temperature kept at 50 degrees? Why was the product stored on ice?
The answers to these questions should be sensible and scientific. Temperatures, amounts and incubation's are used to optimise reactions. Every step should be planned to get the best possible result in the most efficient way.
At the moment, we're growing phages on agar plates. At one stage of the protocol, we use exactly 84 plates to grow them. Two of the plates are controls and four are for dilutions but the fact remains that every time we use exactly 84 plates, no matter what we're doing.
The answer (as can probably be guessed) is not scientific in the least. Science is not a cold and clinical organised space, no matter how much scientists want it to be. It is a wild and crazy world full of human error and things going random and even more human error. The problems are not just scientific, they are also spacial and temporal; incubators are only so big, parts of the lab are only open during certain hours.
And the big jars we use for incubating will only physically fit 42 plates. We've tried to stuff more in but the lids won't shut. And while we have three of them in total the incubator is quite small and only fits two at a time.
42 x 2 = 84
Which wouldn't seem so bad if it weren't for the dilutions and the controls, because once you've put them in there's only room for 39 plates of actual phage sample per jar. 42 is at least a nice round number, but a protocol always looks a bit odd if it starts with the phrase '39 plates were taken...' It begs the question, 'why 39?' and the answer is, scientifically, faintly embarrassing.
Science would be a lot more precise if the world stopped getting in the way.
But far less fun =D
Back to lab Rat-ing
I'M BACK IN THE LAB!
Wow, it's been a while since I've written anything here. Term finally finished (finally!) and now I am back happily being a Lab Rat and currently waiting for media to cool down so I can plate it out. Normally when making agar or other media it just gets melted in the microwave then poured instantly. However at the moment we're using media that needs antibiotics in it after it's been melted, the antibiotics don't like high temperatures so we have to let it cool down a bit first.
Other things I have to do:
This stuff is taking ages to cool down though x|
Wow, it's been a while since I've written anything here. Term finally finished (finally!) and now I am back happily being a Lab Rat and currently waiting for media to cool down so I can plate it out. Normally when making agar or other media it just gets melted in the microwave then poured instantly. However at the moment we're using media that needs antibiotics in it after it's been melted, the antibiotics don't like high temperatures so we have to let it cool down a bit first.
Other things I have to do:
- Read up papers for my project next term (More bacteria! But no phages. Antibiotics instead)
- Write a 3000 word essay about biorefinaries for the end of January
- Revise all the stuff I did this term because I am a lazy Lab Rat and didn't do enough during the term.
- Decide which options I am doing next term and organise my timetable
- Write a CV and pester the person whose Lab I will be working in over summer this time to the point where they will fill in funding forms for me (but not actually get very pissed off at me).
- Write the second chapter of a Very Geeky Story for a friend (it's about proteins...inside a cell...yeah. Currently stars Fos and Jun as the main characters, over-sentimental sacrificial cyclins and a gigantic mafia-style phosphate empire. Like a Sci-Fi/Western. But in a cell. heh)
This stuff is taking ages to cool down though x|
Presenting Histones
Bravery is an interesting word. It's one of those words that has many different subtle shades of definition; ranging from altruism to stupidity. The big question is, of course, was it brave or stupid of me to volunteer to do a presentation at the first supervision of term?
To put this more in context, I haven't actually done a scientific presentation since, well, at all. We did some mini ones in our supervision group last year, but they did not go so well. I haven't had to speak in front of a large crowd of people for about three years, not since the upper sixth performance of 'Dracula' where I stumbled on stage for a few minutes to play Translyvanian Peasant With Godawful Accent.
Here is the title and the link (for those who can get it) for what I have to present:
"Rb targets histone H3 methylation and HP1 to promoters"
I'm going to go through the paper now and try to provide a quick summary of what it is about. I have no idea how I'm meant to present it (hopefully there will be a brief meeting at some point to discuss this) but I can't help but feel things will go slightly better if I actually know what the paper is talking about.
okay... a look at the abstract and one brief scribbled diagram later this is what I've got:
Pretty pictures if you follow the links!
There is lots of DNA in the cell, so in order for it to fit into the nucleus it has to be coiled. One method for coiling involves wrapping the DNA around histone proteins (beads on a string) to keep them coiled. As well as keeping the DNA wound up, histones can also signal to transcription factors (proteins that start the complex process of turning DNA into protein) which bits of the DNA they need to read by displaying chemical signals.
One such signal is the methyl group, -CH3. Sticking a methyl group onto the end of a histone signals to the cells that this DNA is in Do Not Disturb mode, and should not be turned into protein. The study the paper was doing focused on a protein that goes around putting up all the nuclear Do Not Disturb signs; SUV39H1 (which shall henceforth be known as SUVy). This methylated the histones at a certain point (lysine 9 of histone H3 for anyone interested) and keeps the DNA associated with them from being expressed. It does this by recruting HP1 which binds to the DNA and, as far as I can work out from this, just sits there and stops it being expressed.
There are two forms that DNA in the nucleus can take: heterochromatin, which is all coiled up and not doing anything, and enchromatin, which is being actively expressed. This paper was getting fairly excited because while it was known that SUVy and HP1 were good at keeping heterochromatin quiet, they found them interacting with euchromatin! What's more they were consorting with Rb, a very well known protein that is involved in all sorts of processes that supress the expression of DNA, particularly in different parts of the cell cycle.
By doing various assays involving pulling out the Rb bound to DNA and then finding what bit of DNA it was bound to, they discovered that it methylated the same H3 on the lycine that HP1 did. Furthermore, Rb can interact with SUVy, due to a 'pocket domain' which SUVy fits into quite well. The end conclusion of all this is that Rb and SUVy interact together, methylate a part of the DNA which people hadn't really known SUVy was methylating, and then HP1 comes and sits on it.
The exciting thing here (alright not that exciting, but fairly interesting at the least) is that they put forward at the end that there may be other euchromatin repressor proteins out there that bind to SUVy and mobilise the DNA repression in euchromatin. Also, as Rb is involved in cell cycle control, it helps to build a bigger picture of just what is going on in the cell cycle (which cancer reseachers tend to like).
And woohoo I get to do a presentation on it. :)
To put this more in context, I haven't actually done a scientific presentation since, well, at all. We did some mini ones in our supervision group last year, but they did not go so well. I haven't had to speak in front of a large crowd of people for about three years, not since the upper sixth performance of 'Dracula' where I stumbled on stage for a few minutes to play Translyvanian Peasant With Godawful Accent.
Here is the title and the link (for those who can get it) for what I have to present:
"Rb targets histone H3 methylation and HP1 to promoters"
I'm going to go through the paper now and try to provide a quick summary of what it is about. I have no idea how I'm meant to present it (hopefully there will be a brief meeting at some point to discuss this) but I can't help but feel things will go slightly better if I actually know what the paper is talking about.
okay... a look at the abstract and one brief scribbled diagram later this is what I've got:
Pretty pictures if you follow the links!
There is lots of DNA in the cell, so in order for it to fit into the nucleus it has to be coiled. One method for coiling involves wrapping the DNA around histone proteins (beads on a string) to keep them coiled. As well as keeping the DNA wound up, histones can also signal to transcription factors (proteins that start the complex process of turning DNA into protein) which bits of the DNA they need to read by displaying chemical signals.
{kind=link}
One such signal is the methyl group, -CH3. Sticking a methyl group onto the end of a histone signals to the cells that this DNA is in Do Not Disturb mode, and should not be turned into protein. The study the paper was doing focused on a protein that goes around putting up all the nuclear Do Not Disturb signs; SUV39H1 (which shall henceforth be known as SUVy). This methylated the histones at a certain point (lysine 9 of histone H3 for anyone interested) and keeps the DNA associated with them from being expressed. It does this by recruting HP1 which binds to the DNA and, as far as I can work out from this, just sits there and stops it being expressed.
{kind=link}
There are two forms that DNA in the nucleus can take: heterochromatin, which is all coiled up and not doing anything, and enchromatin, which is being actively expressed. This paper was getting fairly excited because while it was known that SUVy and HP1 were good at keeping heterochromatin quiet, they found them interacting with euchromatin! What's more they were consorting with Rb, a very well known protein that is involved in all sorts of processes that supress the expression of DNA, particularly in different parts of the cell cycle.
By doing various assays involving pulling out the Rb bound to DNA and then finding what bit of DNA it was bound to, they discovered that it methylated the same H3 on the lycine that HP1 did. Furthermore, Rb can interact with SUVy, due to a 'pocket domain' which SUVy fits into quite well. The end conclusion of all this is that Rb and SUVy interact together, methylate a part of the DNA which people hadn't really known SUVy was methylating, and then HP1 comes and sits on it.
The exciting thing here (alright not that exciting, but fairly interesting at the least) is that they put forward at the end that there may be other euchromatin repressor proteins out there that bind to SUVy and mobilise the DNA repression in euchromatin. Also, as Rb is involved in cell cycle control, it helps to build a bigger picture of just what is going on in the cell cycle (which cancer reseachers tend to like).
And woohoo I get to do a presentation on it. :)
Proteomics
So, term has started. Lab Rat is out of the lab and back in the lecture theatre, which means probably less blog posts as I try to get through my rather scary reading list (thankfully it is made up mostly of papers but there's still rather a lot of it).
The first topic for this term is Proteomics; the study of the structure and function of proteins within a cell and living proof that adding the term -omics onto something gets you exciting amounts of funding. Proteomics is turning out to be fairly interesting, the paper I've just read, for example (here if you can get to it) talks about how large scale proteomics was used to compare the proteins in the malaria parasite P. falciparum in its different states of growth. The idea is to find a protein in one stage that isn't in the body naturally and then target it to kill off the parasite. Also it's really interesting finding out which proteins are expressed when, and which ones the parasite turns off at different stages for various reasons.
*sigh*
Yeah, I miss phages :(
The first topic for this term is Proteomics; the study of the structure and function of proteins within a cell and living proof that adding the term -omics onto something gets you exciting amounts of funding. Proteomics is turning out to be fairly interesting, the paper I've just read, for example (here if you can get to it) talks about how large scale proteomics was used to compare the proteins in the malaria parasite P. falciparum in its different states of growth. The idea is to find a protein in one stage that isn't in the body naturally and then target it to kill off the parasite. Also it's really interesting finding out which proteins are expressed when, and which ones the parasite turns off at different stages for various reasons.
*sigh*
Yeah, I miss phages :(
species specific mouse transcription and the start of term
Term is about to start! I still can't believe that the holiday is almost over :( On the other hand, this holiday does seem to have gone on a while, memories of exams and lectures and learning seem so far away.
I will have to step away from being a Lab Rat for a while, and go back to being a Student. Lectures, reading, more reading, desperately understanding and (a new one for this year) Seminars. Seminars are where some other lab rat stands at the front of the room and talks about their lab ratting for a bit and then everyone else has to try to think of clever things to say about it. It's all great fun and sometimes you get free wine or food.
So as it's the start of term and I'm full of Good Intentions I went over to my departmental webpage to see if any of the abstracts for the seminars were up. It turns out that the first one is up. Here it is:
"Homologous sets of transcription factors direct conserved tissue-specific gene expression, yet transcription factor binding events diverge rapidly between closely related species. We used hepatocytes from an aneuploid mouse strain carrying human chromosome 21 to determine on a chromosomal scale whether interspecies differences in transcriptional regulation are primarily directed by human genetic sequence or mouse nuclear environment. Virtually all transcription factor binding locations, landmarks of transcription initiation, and the resulting gene expression observed in human hepatocytes were recapitulated across the entire human chromosome 21 in the mouse hepatocyte nucleus. Thus, in homologous tissues, genetic sequence is largely responsible for directing transcriptional programs; interspecies differences in epigenetic machinery, cellular environment, and transcription factors themselves play secondary roles."
I actually almost fainted when I read that. A few deep breaths later and I decided to come back to it and understand it. (Why can't scientists write what they mean?)
Translation (if you want to skip my waffle and get straight to a quick translation skip to the italic bit)
I have to admit that I had a bit of a clue to help with the translation, they gave me the guys webpage. A quick look confirmed that he works for cancer research UK, which helps because there are actually a limited number of things people in cancer research tend to work with (well, at the very least it confirms that he's not working with bacteria...)
So...the first sentence. Transcription factors are proteins that bind to the DNA and control its expression. Probably a term most biochemists should really know (heh). Homologous just means 'pretty much the same'. So their problem is that they've got transcription factors in very similar species doing wildly different things.
Hepatocytes are liver cells. Aneuploidy means 'an abnormal number of chromosomes'. So they basically just stuck human chromosome 21 (the smallest one! and, incidentally, the one that leads to Down syndrome) into mouse liver cells to see what they did. The idea being to find out whether the differences were caused by the actual nuclear material or the 'epigenetic' surroundings (epigenetic = stuff that isn't DNA)
Epigenetics is a relatively new and exciting concept incidentally. It's the idea that the actual nuclear environment has a large part to play in what gets expressed rather than just the DNA as much. Also it's an idea that really pisses off James Watson and anything that pisses off James Watson is fine by me.
So what happened? Were the alien chromosome 21's expressed like mouse chromosomes (showing epigenetic control) or exactly as they would be in humans (showing DNA control). The answer is in the third sentence, the chromosome 21's were expressed exactly the same as they were in humans. No epigenetics here :(
*these aren't the epigenetic controls you're looking for*
For those who just want a simple translation here it is, to the best of my ability:
Similar sets of transcription factors are involved in controlling the expression of mouse DNA. However the way they work is very different, even in closely related species. We placed the human chromosome 21 into mouse liver cells to see if they were expressed like mouse chromosomes (showing control from factors other than the DNA) or like human chromosomes (showing that all control is from the DNA). The expression of the chromosome 21 in the mouse liver cell nucleus was almost identical to the way it is expressed in humans. Therefore, in similar tissues, it is the genetic sequence that determines how the DNA is expressed, all other factors are secondary.
Now I've got to try and think of clever questions to ask about that. I might have a try at getting hold of the paper, then at the very least I can ask poncy questions about the techniques.
I will have to step away from being a Lab Rat for a while, and go back to being a Student. Lectures, reading, more reading, desperately understanding and (a new one for this year) Seminars. Seminars are where some other lab rat stands at the front of the room and talks about their lab ratting for a bit and then everyone else has to try to think of clever things to say about it. It's all great fun and sometimes you get free wine or food.
So as it's the start of term and I'm full of Good Intentions I went over to my departmental webpage to see if any of the abstracts for the seminars were up. It turns out that the first one is up. Here it is:
"Homologous sets of transcription factors direct conserved tissue-specific gene expression, yet transcription factor binding events diverge rapidly between closely related species. We used hepatocytes from an aneuploid mouse strain carrying human chromosome 21 to determine on a chromosomal scale whether interspecies differences in transcriptional regulation are primarily directed by human genetic sequence or mouse nuclear environment. Virtually all transcription factor binding locations, landmarks of transcription initiation, and the resulting gene expression observed in human hepatocytes were recapitulated across the entire human chromosome 21 in the mouse hepatocyte nucleus. Thus, in homologous tissues, genetic sequence is largely responsible for directing transcriptional programs; interspecies differences in epigenetic machinery, cellular environment, and transcription factors themselves play secondary roles."
I actually almost fainted when I read that. A few deep breaths later and I decided to come back to it and understand it. (Why can't scientists write what they mean?)
Translation (if you want to skip my waffle and get straight to a quick translation skip to the italic bit)
I have to admit that I had a bit of a clue to help with the translation, they gave me the guys webpage. A quick look confirmed that he works for cancer research UK, which helps because there are actually a limited number of things people in cancer research tend to work with (well, at the very least it confirms that he's not working with bacteria...)
So...the first sentence. Transcription factors are proteins that bind to the DNA and control its expression. Probably a term most biochemists should really know (heh). Homologous just means 'pretty much the same'. So their problem is that they've got transcription factors in very similar species doing wildly different things.
Hepatocytes are liver cells. Aneuploidy means 'an abnormal number of chromosomes'. So they basically just stuck human chromosome 21 (the smallest one! and, incidentally, the one that leads to Down syndrome) into mouse liver cells to see what they did. The idea being to find out whether the differences were caused by the actual nuclear material or the 'epigenetic' surroundings (epigenetic = stuff that isn't DNA)
{kind=link}
Epigenetics is a relatively new and exciting concept incidentally. It's the idea that the actual nuclear environment has a large part to play in what gets expressed rather than just the DNA as much. Also it's an idea that really pisses off James Watson and anything that pisses off James Watson is fine by me.
So what happened? Were the alien chromosome 21's expressed like mouse chromosomes (showing epigenetic control) or exactly as they would be in humans (showing DNA control). The answer is in the third sentence, the chromosome 21's were expressed exactly the same as they were in humans. No epigenetics here :(
*these aren't the epigenetic controls you're looking for*
For those who just want a simple translation here it is, to the best of my ability:
Similar sets of transcription factors are involved in controlling the expression of mouse DNA. However the way they work is very different, even in closely related species. We placed the human chromosome 21 into mouse liver cells to see if they were expressed like mouse chromosomes (showing control from factors other than the DNA) or like human chromosomes (showing that all control is from the DNA). The expression of the chromosome 21 in the mouse liver cell nucleus was almost identical to the way it is expressed in humans. Therefore, in similar tissues, it is the genetic sequence that determines how the DNA is expressed, all other factors are secondary.
Now I've got to try and think of clever questions to ask about that. I might have a try at getting hold of the paper, then at the very least I can ask poncy questions about the techniques.
Taxonomists vs. Phages
Taxonomy is one of those wonderful subjects that at first seems very simple (and very boring). The word comes from Greek - taxis meaning 'order' and nomos meaning 'law', or 'science' and is at it's most basic the science of classification. Most people do varying amounts of taxonomy at school; dividing living things into plants, animals and fungi, dividing the vertebrates into birds, mammals, amphibians, fish and reptiles etc.
Putting the dubious accuracy value of these classification labels aside (particularly the bit about reptiles) taxonomy is, at it's most basic level, a simple system for ordering things and putting them in little boxes. Glorified stamp collecting, as it were. But there are still plenty of arguments and various feuds about the exact relationships of things, most often at the species level, and even whether the whole 'five kingdom' model is any use (the five kingdoms are animals, plants, fungi, prokaryotes - roughly bacteria, and protoctista - which are basically the equivalent of the filing draw marked misc.). In fact, the more you delve into the science of taxonomy, the more complications and problems you start to encounter. Even something that would seem fairly simple, such as what defines a 'species' is a matter of hot debate.
And when you get down to the level of single-celled organisms the whole system goes a bit haywire. The distinction between 'animals', 'plants' and 'fungi' completely breaks down, there are single-celled things that are motile and animal-like but photosynthesise, things that only photosynthesise at the right time of day, or are perfectly sessile and plant-like except they don't photosynthesise. Most protozoa (single celled thingys, more information under the link) are now broken up into a whole new set of labels, very few of which seem to relate to the larger multi-cellular organisms.
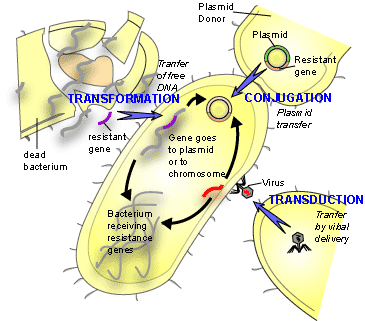
When it gets to things like bacteria and bacteriophages, of course, taxonomists just break down and cry. Because bacteria, unlike most other things, don't even maintain their genetic integrity. Bacteria can share bits of their genome quite happily, even with bacteria that are seemingly very unrelated, making the whole 'species' concept break down a bit anyway. Phages merrily incorporate various bits of bacteria into themselves, splice bits out, even splice themselves in to bacterial genomes and sit there for a while. It's a complete headache to try and organise the things.
Various attempts have been made, of course, since the first discovery of phages in 1915 by the wonderfully python-esquely named Frederick Twort. (to give all and full credit they were also discovered completely independently in 1917 by Felix d'Herelle). The first system was based on morphology, what the phage looked like, and was greatly helped by the electron microscope. Although most phages adopt the 'lunar landing module' look, there is plenty of variation within that. Length of tail fibres, size of capsid, symmetry of the capsule, alright, not very much variation, but still something for taxonomists to hang onto.
Size and shape are never good indications of relatedness, a dolphin is more related to a hyena than to a shark, however similar the two might look. Nucleic acid research during the 1960's shook up the whole discipline of taxonomy by providing lots of new exciting DNA information. Phages could now be classified by the amount and type of DNA, which, added to the morphological data, provided a system (albeit a slightly wobbly one).
One of the most currently most widely popular methods to classify things is to examine the genetic DNA that codes for the ribosomes. Ribosomes are complexes of RNA and protein that are used to turn the genetic code into proteins. They are very highly conserved and are therefore very useful in determining evolutionary relatedness and taxonomy.
The problem is, of course, that bacteriophages don't have any ribosomes. They use the bacterial ones; they harness the bacterial internal machinery for replicating DNA and making proteins and therefore don't need to carry any of their own. In view of this, one of the most recent attempts to organise phage taxonomy has focused on looking at their proteins. The relatedness of the proteins has been used to create clever sounding things like distance matrices and the highly impressively named 'phage proteome tree'. Of course there are several problems, possibly the main one being that phages, especially the ones that sit inside bacteria, have a distressing tendency to pick up bits of DNA that aren't theirs. Which translates into proteins that aren't theirs and makes the whole procedure just that little bit more awkward.
There's been some work comparing the genomes of just the structural components, in the hope that there won't be too much genetic exchange going on with the genes that are actually needed to build the phage. The people doing it seem fairly confident, and have managed to isolate about five separate genera. There's a paper from them here, hopefully should be accessible (in form if not in content).
The whole thing is really all a bit up in the air, with some fairly amusing piss-ups between the different schools of thought. Horizontal gene transfer can be a bitch sometimes :)
Putting the dubious accuracy value of these classification labels aside (particularly the bit about reptiles) taxonomy is, at it's most basic level, a simple system for ordering things and putting them in little boxes. Glorified stamp collecting, as it were. But there are still plenty of arguments and various feuds about the exact relationships of things, most often at the species level, and even whether the whole 'five kingdom' model is any use (the five kingdoms are animals, plants, fungi, prokaryotes - roughly bacteria, and protoctista - which are basically the equivalent of the filing draw marked misc.). In fact, the more you delve into the science of taxonomy, the more complications and problems you start to encounter. Even something that would seem fairly simple, such as what defines a 'species' is a matter of hot debate.
And when you get down to the level of single-celled organisms the whole system goes a bit haywire. The distinction between 'animals', 'plants' and 'fungi' completely breaks down, there are single-celled things that are motile and animal-like but photosynthesise, things that only photosynthesise at the right time of day, or are perfectly sessile and plant-like except they don't photosynthesise. Most protozoa (single celled thingys, more information under the link) are now broken up into a whole new set of labels, very few of which seem to relate to the larger multi-cellular organisms.
When it gets to things like bacteria and bacteriophages, of course, taxonomists just break down and cry. Because bacteria, unlike most other things, don't even maintain their genetic integrity. Bacteria can share bits of their genome quite happily, even with bacteria that are seemingly very unrelated, making the whole 'species' concept break down a bit anyway. Phages merrily incorporate various bits of bacteria into themselves, splice bits out, even splice themselves in to bacterial genomes and sit there for a while. It's a complete headache to try and organise the things.
{kind=link}
Various attempts have been made, of course, since the first discovery of phages in 1915 by the wonderfully python-esquely named Frederick Twort. (to give all and full credit they were also discovered completely independently in 1917 by Felix d'Herelle). The first system was based on morphology, what the phage looked like, and was greatly helped by the electron microscope. Although most phages adopt the 'lunar landing module' look, there is plenty of variation within that. Length of tail fibres, size of capsid, symmetry of the capsule, alright, not very much variation, but still something for taxonomists to hang onto.
{kind=link}
Size and shape are never good indications of relatedness, a dolphin is more related to a hyena than to a shark, however similar the two might look. Nucleic acid research during the 1960's shook up the whole discipline of taxonomy by providing lots of new exciting DNA information. Phages could now be classified by the amount and type of DNA, which, added to the morphological data, provided a system (albeit a slightly wobbly one).
One of the most currently most widely popular methods to classify things is to examine the genetic DNA that codes for the ribosomes. Ribosomes are complexes of RNA and protein that are used to turn the genetic code into proteins. They are very highly conserved and are therefore very useful in determining evolutionary relatedness and taxonomy.
The problem is, of course, that bacteriophages don't have any ribosomes. They use the bacterial ones; they harness the bacterial internal machinery for replicating DNA and making proteins and therefore don't need to carry any of their own. In view of this, one of the most recent attempts to organise phage taxonomy has focused on looking at their proteins. The relatedness of the proteins has been used to create clever sounding things like distance matrices and the highly impressively named 'phage proteome tree'. Of course there are several problems, possibly the main one being that phages, especially the ones that sit inside bacteria, have a distressing tendency to pick up bits of DNA that aren't theirs. Which translates into proteins that aren't theirs and makes the whole procedure just that little bit more awkward.
There's been some work comparing the genomes of just the structural components, in the hope that there won't be too much genetic exchange going on with the genes that are actually needed to build the phage. The people doing it seem fairly confident, and have managed to isolate about five separate genera. There's a paper from them here, hopefully should be accessible (in form if not in content).
The whole thing is really all a bit up in the air, with some fairly amusing piss-ups between the different schools of thought. Horizontal gene transfer can be a bitch sometimes :)
What to do with DNA
Now we've actually managed to extract our DNA (like this) we get to cut it all up into tiny pieces. It might seem a bit pointless, but this is how to determine that we actually have the right bits of DNA in our extracts. Cutting DNA with little enzymes called restriction enzymes produces different restriction patters depending on what DNA you have.
Restriction enzymes are naturally produced by most bacteria, and what they do is cut pieces of DNA at very specific points. EcoRI, for example, cuts DNA after the G in the DNA sequence GAATTC. As each viral genome has a different DNA sequence, each one will produce a different restriction map, producing a characteristic number of bands on a gel:
 (this picture is not from my research, it is from here)
(this picture is not from my research, it is from here)
Each band is a blob of a certain size of DNA lit up with ethidium bromide (which is a dye, nothing very exotic). Different restriction enzymes, and different genomes, will produce different band patterns on the gel.
So, what do the bacteria need to produce DNA cutting enzymes for? The answer (naturally) is bacteriophages! One way the bacteria can protect themselves against viral invasion is to have lots of these enzymes around. As soon as the viruses inject their DNA into the bacteria cell, the restriction enzymes chop it all up.
But bacteria also contain DNA, and unlike people (and other eukaryotes), they don't keep it all tucked up in a nuclear membrane. So how do they stop the restriction enzymes from cutting up their DNA? One of the most common ways is to methylate the DNA, essentially sticking a methyl group (a carbon atom attached to three hydrogen atoms) onto some of the bases. in the example shown above, therefore, the restriction enzyme is looking for the sequence GAATTC. It sees this in invading DNA and slices it up, but in the bacterias own DNA it sees GA(methylated)A(methylated)TTC, which it doesn't recognise. And therefore, does not cut.
Restriction enzymes were first discovered my Daniel Nathans, Werner Arber, and Hamilton Smith. They won the Nobel Prize for it in 1978. (see here)
Restriction enzymes are naturally produced by most bacteria, and what they do is cut pieces of DNA at very specific points. EcoRI, for example, cuts DNA after the G in the DNA sequence GAATTC. As each viral genome has a different DNA sequence, each one will produce a different restriction map, producing a characteristic number of bands on a gel:
 (this picture is not from my research, it is from here)
(this picture is not from my research, it is from here){kind=link}
Each band is a blob of a certain size of DNA lit up with ethidium bromide (which is a dye, nothing very exotic). Different restriction enzymes, and different genomes, will produce different band patterns on the gel.
So, what do the bacteria need to produce DNA cutting enzymes for? The answer (naturally) is bacteriophages! One way the bacteria can protect themselves against viral invasion is to have lots of these enzymes around. As soon as the viruses inject their DNA into the bacteria cell, the restriction enzymes chop it all up.
But bacteria also contain DNA, and unlike people (and other eukaryotes), they don't keep it all tucked up in a nuclear membrane. So how do they stop the restriction enzymes from cutting up their DNA? One of the most common ways is to methylate the DNA, essentially sticking a methyl group (a carbon atom attached to three hydrogen atoms) onto some of the bases. in the example shown above, therefore, the restriction enzyme is looking for the sequence GAATTC. It sees this in invading DNA and slices it up, but in the bacterias own DNA it sees GA(methylated)A(methylated)TTC, which it doesn't recognise. And therefore, does not cut.
{kind=link}
Restriction enzymes were first discovered my Daniel Nathans, Werner Arber, and Hamilton Smith. They won the Nobel Prize for it in 1978. (see here)
Writing in officialese
One of the things about science nowadays is that it tends to generate impressive amounts of paperwork, which you have to wade through to get to the actual science. For example, I want to keep working as a Lab Rat to help out my supervisor. The money is available to fund me, but I can't actually get at it without filling out a little form explaining why.
It isn't too bad, two sides of A4 with mostly just information about who I am and what my details are. There's only one part where I have to do any actual writing, so I'm currently trying to figure out how to say "I want to work in a lab! It is fun! It will give me CV points!" in officialese. I have a feeling that writing "I want something on my CV. Duh, why do you think I want to work in September?" would probably be frowned upon. As would seeming excessively keen. I don't know though, is it acceptable to write that you actually enjoy lab work on a form? Or will they just think I'm making it up.
It's all good practise though. Sticking with lab work means that my future will be full of funding forms and various other bits of paperwork in whch I try to find convincing and acceptible reasons for doing what I do. And then trying to couch them in slightly better terms than "I want money. I like lab work. Give me money, I will give you work."
heh. It's like applying to university all over again. ("As well as achieving impressive exam results I have had lots of experience doing all sorts of intelligent things-FOR THE LOVE OF ALL THINGS HOLY JUST SOMEONE LET ME IN")
It isn't too bad, two sides of A4 with mostly just information about who I am and what my details are. There's only one part where I have to do any actual writing, so I'm currently trying to figure out how to say "I want to work in a lab! It is fun! It will give me CV points!" in officialese. I have a feeling that writing "I want something on my CV. Duh, why do you think I want to work in September?" would probably be frowned upon. As would seeming excessively keen. I don't know though, is it acceptable to write that you actually enjoy lab work on a form? Or will they just think I'm making it up.
It's all good practise though. Sticking with lab work means that my future will be full of funding forms and various other bits of paperwork in whch I try to find convincing and acceptible reasons for doing what I do. And then trying to couch them in slightly better terms than "I want money. I like lab work. Give me money, I will give you work."
heh. It's like applying to university all over again. ("As well as achieving impressive exam results I have had lots of experience doing all sorts of intelligent things-FOR THE LOVE OF ALL THINGS HOLY JUST SOMEONE LET ME IN")
sometimes life gets you like that
This was going to be quite a long soul-searching post about the general usefulness of science, based on a conversation held the other evening about whether the Large Hadron Collider was actually worth it. Huge amounts of time, money and energy (and a slight amount of danger) all for ... what? The Higgs partical? Why? Seriously, is there any point to finding this thing except just ... because?
But I am tired, and vaguely upset, and really not in the mood to spend a goodly amount of time talking about how the subject I ended up doing (and will keep on doing) is a mighty large pile of intellectual w*nk. It is starting to look very definitely like science is going to constitute a large part of my life, and I need to be a lot more awake and cheerful before pulling my life apart like that.
So I'm going to read firefly fanfiction instead.
Literature degree? Should'a could'a would'a.
But I am tired, and vaguely upset, and really not in the mood to spend a goodly amount of time talking about how the subject I ended up doing (and will keep on doing) is a mighty large pile of intellectual w*nk. It is starting to look very definitely like science is going to constitute a large part of my life, and I need to be a lot more awake and cheerful before pulling my life apart like that.
So I'm going to read firefly fanfiction instead.
Literature degree? Should'a could'a would'a.
Not quite the end of the world...
The Large Hadron Collider has just been turned on! I should probably be slightly more excited by this, but I am slightly upset today due to the devistating discovery that our mutant DNA isn't. We have lots of lovely wild-type DNA and no mutant strains at all. *sigh*
But anyway, the Hadron Collider. As yet, no black holes have opened, and no aleins or stragelets have appeared. Not particularly surprising though, as as far as I can understand, switching it on was never the issue, the problem was with the actual collisions. Which haven't started yet. So far, they've just been shooting protons around the circit, in a clockwise direction. Later, they're going to start sending them anticlockwise as well. The actual high-energy collisions aren't due to start until the 21st October.
For the record, I'm fairly sure nothing particularly bad will happen when they do start the collisions. Hopefully they'll find something interesting, they may even find something useful. And if it all does go horrendously wrong, it is a slightly comforting thought that we won't actually know about it.
(Although if it creates a black hole that sucks the whole universe up my tentative plans for reincarnation might end up severly scuppered).
But anyway, the Hadron Collider. As yet, no black holes have opened, and no aleins or stragelets have appeared. Not particularly surprising though, as as far as I can understand, switching it on was never the issue, the problem was with the actual collisions. Which haven't started yet. So far, they've just been shooting protons around the circit, in a clockwise direction. Later, they're going to start sending them anticlockwise as well. The actual high-energy collisions aren't due to start until the 21st October.
For the record, I'm fairly sure nothing particularly bad will happen when they do start the collisions. Hopefully they'll find something interesting, they may even find something useful. And if it all does go horrendously wrong, it is a slightly comforting thought that we won't actually know about it.
(Although if it creates a black hole that sucks the whole universe up my tentative plans for reincarnation might end up severly scuppered).
Reflective Learning
The end of my project is coming up unfortunately (although I still get to stay in the lab so wo0t, not too bad) and one of the things I should probably do for my portfolio is a short 'reflective learning' sheet. i.e, What I Have Gained From This Experience.
The short answer is: A lot. I have enjoyed (almost) every minute of lab work, it's been frustrating at times, sure, but it's basically just been one hell of an awesome ride. The thing is, in my official 'reflective learning' thing I should probably focus on things of Practical Value. Various techniques and things I have picked up, information I have learned about working in a lab environment, an increased awareness of the workings of science etc.
In reality, of course, the things I've actually picked up are far vaguer and more interesting. So here is the unofficial version of what I've really got out of the whole experience. They don't tick any boxes in forms, but they are somehow a lot more important:
And hopefully that attitude will stay with me throughout next term, and encourage me to actually work hard :)
The short answer is: A lot. I have enjoyed (almost) every minute of lab work, it's been frustrating at times, sure, but it's basically just been one hell of an awesome ride. The thing is, in my official 'reflective learning' thing I should probably focus on things of Practical Value. Various techniques and things I have picked up, information I have learned about working in a lab environment, an increased awareness of the workings of science etc.
In reality, of course, the things I've actually picked up are far vaguer and more interesting. So here is the unofficial version of what I've really got out of the whole experience. They don't tick any boxes in forms, but they are somehow a lot more important:
- Reflexes. I've gained a whole lot more reflexes and instinctive responses, to a vaguely Pavlovian turn of the head when an alarm goes off to a vital spacial awareness of where the end of a pipette tip is.
- What happens in a lab. Mostly washing up and cookery. The science comes in at the beginning when you write a protocol and at the end when you stare in confusion at your results. The bit in the middle is mostly cookery.
- Organisation. Oh ghod. Probably the best thing I've got from this is the beginnings of development of a healthy paranoia about labelling things. Label and date everything, even with the useless information.
- Small writing. I am getting very good at writing tiny labels on miniature eppindorfs.
- Pragmatism. Sometimes experiments work. Sometimes they don't. Life is not predictable. The lab may be scientific, but the organisms damn well aren't. That's how it is. Squint at the protocol, get new equipment, shrug, and do the whole thing all over. (and if it works, you cna spin round on your chair making sqeaky noises)
- Temporal awareness. Everything takes longer than you think. Everything.
- Orders of magnitude. Never underestimate the ability of an order of magnitude to suddenly vanish. There is a big difference between 10 and 100, which has a tendency to disappear at crucial times.
{kind=link}
And hopefully that attitude will stay with me throughout next term, and encourage me to actually work hard :)
Patron Saints
So during a particularly long session, with fairly crucial results, I did a bit of a google to see if there was anyone that the Catholic church, in its infinate wisdom, had put aside to recieve the various prayers and obscinities coming from those persuing the noble discipline of science. It turns out (interestingly enough) that there is:

The rather imposing man pictured above is Saint Albert the Great (1206-1280). The official Patron Saint of science. Also known as (according to saints.SPQN.com) Albertus Magnus, Doctor Expertus and Doctor Universalis. My hazy grasp of Latin seems to suggest the last one may be a tad over the top for philosophy teacher who liked browsing science books but there you go. To my amazement, there is also a little prayer to say to him:

Thomas Aquinus, patron saint of students and academics. A quick bit of googling also confirms that he is the patron saint against storms, against lightning, of apologists, of chastity and (for reasons best known to himself) of pencil-makers. He has a lot more alternative names as well: Angelic Doctor, Doctor Angelicus (the same thing but in Latin, surely?), Doctor Communis, Great Synthesizer (there should be something funny to say about this one, can't think of it though), The Universal Teacher and, rather bizarrely, The Dumb Ox.
I love the catholic Saints. They are like the old polytheistic gods (except a lot less interesting) and there seems to be one for almost anything you care to think of.

The rather imposing man pictured above is Saint Albert the Great (1206-1280). The official Patron Saint of science. Also known as (according to saints.SPQN.com) Albertus Magnus, Doctor Expertus and Doctor Universalis. My hazy grasp of Latin seems to suggest the last one may be a tad over the top for philosophy teacher who liked browsing science books but there you go. To my amazement, there is also a little prayer to say to him:
Dear Scientist and Doctor of the Church, natural science always led you to the higher science of God. Though you had an encyclopedic knowledge, it never made you proud, for you regarded it as a gift of God. Inspire scientists to use their gifts well in studying the wonders of creation, thus bettering the lot of the human race and rendering greater glory to God. Amen
For someone known as the 'Doctor Universalis' I'm not entirely sure if the 'never made you proud' bit scans wonderfully well. Also the prayer is not as specific as could be hoped. Albert the Great make this bloody gel work feels slightly more appropriate. And while my phages may hopefully one day end up bettering the human race (heh) I'm not entirely sure how they bring glory to god (would phages be able to pray, if taught? Could they destroy bacteria in a particularly holy way?). I am quite surprised though, that he turned up, I wasn't expecting there to be anyone specific for science, if anything I was expecting this guy:

Thomas Aquinus, patron saint of students and academics. A quick bit of googling also confirms that he is the patron saint against storms, against lightning, of apologists, of chastity and (for reasons best known to himself) of pencil-makers. He has a lot more alternative names as well: Angelic Doctor, Doctor Angelicus (the same thing but in Latin, surely?), Doctor Communis, Great Synthesizer (there should be something funny to say about this one, can't think of it though), The Universal Teacher and, rather bizarrely, The Dumb Ox.
I love the catholic Saints. They are like the old polytheistic gods (except a lot less interesting) and there seems to be one for almost anything you care to think of.
On the specificity of phages
One of the main objections that I have heard to the use of bacteriophages as therapeutic agents is that phages tend to be very very specific. Each phage usually only attacks one type of bacteria, e.g a bacteriophage for a certain type of e. coli will usually only attack that specific e. coli type and no other bacteria. As there are a large number of different varieties of e. coli (and indeed many other pathogenic bacteria), this specificity could be a problem for targeting infections.
(as an aside, phages are often so specific that they are used to 'type' bacteria. If you find a bacteria in, say, sewage, one way to find out what it is is to attack it with different phages and see which one digests it. This can give a very specific pinpoint of what bacteria you have).
The usual response to the problem of phage specificity is to suggest that a cocktail of phages can be used, one for each potential type of attacking bacteria. In some ways, this can be even more useful for controlling infection; a broad-spectrum antibiotic will knock out any bacteria it comes across while a cocktail of phages will target specifically the unwanted ones. Going back to the example of e.coli: there are lots of e. coli living happily in your gut. If a pathogenic strain gets in, the phage therapy that you are given could be designed to target just the pathogens, rather than the bacteria that are already there (and are necessary for correct digestion).
I was quite surprised therefore to come across this paper while randomly searching PubMed (as you do). Maddeningly, there seems to be no way to get the the actual paper, but what it says is that a phage has been found (and named KVP40 for those interested) which has quite a wide host range. Not only does it attack a variety of Vibrio phages (both pathogenic and non-pathogenic) it also is able to attack a Photobacterium as well (Photobacterium leiognathi). A further paper states in the introduction that the receptor that the Vibrio uses to bind to the surface of its bacterial hosts is the OmpK outer membrane protein. I am not entirely certain what this protein does, but I have found that it is involved in vibrio bacterium immunoprotection and is also present in the photobacterium species. As it elicits a large immune response, it is also being considered as a vaccination, if not in people then at least in the yellow croaker (which is a fish).
And this is where phage therapy could come in useful. I don't know how dangerous the vibrio phage is in yellow croakers (this paper seems to make quite a thing of it, but that may be linked to funding purposes). With enough commercial interest (if it hasn't already happened), it's only a matter of time before someone starts looking for an antibiotic for Vibrio harveyi, the vibio species that attacks the poor fish. Essentially this means that a large amount of time and effort will be spent looking for something that will attack the vibrio species, find it on the basis of the OmpK protein, and then destroy it.
Completely ignoring, of course, the fact that such a thing already exists, in the form of the KVP40 vibriophage. There are millions of them floating around in the sea. They've been isolated as well, in pure phage form.
More people need to be working on this stuff...
(as an aside, phages are often so specific that they are used to 'type' bacteria. If you find a bacteria in, say, sewage, one way to find out what it is is to attack it with different phages and see which one digests it. This can give a very specific pinpoint of what bacteria you have).
The usual response to the problem of phage specificity is to suggest that a cocktail of phages can be used, one for each potential type of attacking bacteria. In some ways, this can be even more useful for controlling infection; a broad-spectrum antibiotic will knock out any bacteria it comes across while a cocktail of phages will target specifically the unwanted ones. Going back to the example of e.coli: there are lots of e. coli living happily in your gut. If a pathogenic strain gets in, the phage therapy that you are given could be designed to target just the pathogens, rather than the bacteria that are already there (and are necessary for correct digestion).
I was quite surprised therefore to come across this paper while randomly searching PubMed (as you do). Maddeningly, there seems to be no way to get the the actual paper, but what it says is that a phage has been found (and named KVP40 for those interested) which has quite a wide host range. Not only does it attack a variety of Vibrio phages (both pathogenic and non-pathogenic) it also is able to attack a Photobacterium as well (Photobacterium leiognathi). A further paper states in the introduction that the receptor that the Vibrio uses to bind to the surface of its bacterial hosts is the OmpK outer membrane protein. I am not entirely certain what this protein does, but I have found that it is involved in vibrio bacterium immunoprotection and is also present in the photobacterium species. As it elicits a large immune response, it is also being considered as a vaccination, if not in people then at least in the yellow croaker (which is a fish).
{kind=link}
And this is where phage therapy could come in useful. I don't know how dangerous the vibrio phage is in yellow croakers (this paper seems to make quite a thing of it, but that may be linked to funding purposes). With enough commercial interest (if it hasn't already happened), it's only a matter of time before someone starts looking for an antibiotic for Vibrio harveyi, the vibio species that attacks the poor fish. Essentially this means that a large amount of time and effort will be spent looking for something that will attack the vibrio species, find it on the basis of the OmpK protein, and then destroy it.
Completely ignoring, of course, the fact that such a thing already exists, in the form of the KVP40 vibriophage. There are millions of them floating around in the sea. They've been isolated as well, in pure phage form.
More people need to be working on this stuff...
Speaking to computers
I am a Lab Rat. I work in laboratories, I try to figure out gels and results. I pipette tiny amounts of liquid into various locations. I try to see patterns and shapes and fit things together. This is what I do. I also occasionally venture into the world of Literature and try to find patterns there (but only as a hobby unfortunately).
What I don't do is computers. I can think of over twenty ways to re-phrase the instruction 'look for the comma' (probably over thirty ways if I'm allowed to use the 'synonyms' feature in word) but not one of those ways works for a computer.
Unfortunately there are some things that even Lab Rats need computers for. For instance, searching for a particular domain of DNA, pulling all the results out of a standard BLAST search, and then taking only the relevant information from that. To slightly clarify, BLAST is a bioinformatics programme with a huge database of information about every known and officially sequenced protein and DNA sequence. You type in your sequence (or your name, if you feel bored) and it shows you what proteins it matches on the database. Unfortunately it provides quite a large amount of information about each one, so this is where programming comes in, you tell the computer which bits of the database you want.
Now I did do computer science for IGCSE. I know what pseudocode is, and how to use it, and could probably stab a guess at setting commands up in the right sequence as well. Where I fall apart is the bit after that, translating the pseudocode into computer speak. There are some phrases I quite literally cannot do.
e.g:
IF comma is present
THEN stop
concatenate new information to old (concatenate=attach or add to)
OK. Fine. That works. But how do you say 'comma is present' in computer? The comma is not equal to anything, so that's out. Nor is the comma in relation to anything, it's just a comma in the middle of the string of writing. I have no idea at all how to tell the computer to find me a comma, Perl (which I am using to programme) seems to have no random squiggle that means 'find' or 'look for'.
Another thing that flummoxed me was the concatenation. All the online tutorials showed you exactly how to concatenate, but only if you already knew what the phrases were:
Tutorial: To concatenate x and y type x.=y
Lab Rat: I DON'T KNOW WHAT X AND Y ARE!!! In fact I'm looking for them because I don't know what they are. I want to find that out!;
Tutorial: *is no help at all*
Lab Rat: *Kicks computer, then hold up a large picture of a comma in front of the screen* Just find this, OK? See this picture, find something that looks like this and then give all the writing in front of it to me;
(the semicolon is computer for 'end of line'. I do not know why computers do this when almost every living person uses a full stop).
Computer: *Is not impressed*
I did actually get there in the end, to the surprise and delight of both myself and my supervisor (and probably the computer as well) I managed to get it to do sort of what I wanted. Unfortunately when we looked back over the raw data from our BLAST results we realised that there was a lot more information we wanted, so a lot more code has to be written. And here we hit another problem. The BLAST databases are truly amazing but just not particularly well organised. Some of them have the important information stored under /notes, while others have a separate field called /function. One item we saw even had the full protein function listed under /name. This means that to get all the information we need, we'll have to pull out each of these fields for every single protein, which will provide us with a lot of useless notes that we don't really need.
Lab Rat: Just give me the useful stuff, OK?;
Computer: Variable 'useful' not defined. Random computer squiggles, out of cheese error.
Lab Rat: *gives up on computers*;
Computer: *gives up on Lab Rat*
What I don't do is computers. I can think of over twenty ways to re-phrase the instruction 'look for the comma' (probably over thirty ways if I'm allowed to use the 'synonyms' feature in word) but not one of those ways works for a computer.
Unfortunately there are some things that even Lab Rats need computers for. For instance, searching for a particular domain of DNA, pulling all the results out of a standard BLAST search, and then taking only the relevant information from that. To slightly clarify, BLAST is a bioinformatics programme with a huge database of information about every known and officially sequenced protein and DNA sequence. You type in your sequence (or your name, if you feel bored) and it shows you what proteins it matches on the database. Unfortunately it provides quite a large amount of information about each one, so this is where programming comes in, you tell the computer which bits of the database you want.
Now I did do computer science for IGCSE. I know what pseudocode is, and how to use it, and could probably stab a guess at setting commands up in the right sequence as well. Where I fall apart is the bit after that, translating the pseudocode into computer speak. There are some phrases I quite literally cannot do.
e.g:
IF comma is present
THEN stop
concatenate new information to old (concatenate=attach or add to)
OK. Fine. That works. But how do you say 'comma is present' in computer? The comma is not equal to anything, so that's out. Nor is the comma in relation to anything, it's just a comma in the middle of the string of writing. I have no idea at all how to tell the computer to find me a comma, Perl (which I am using to programme) seems to have no random squiggle that means 'find' or 'look for'.
Another thing that flummoxed me was the concatenation. All the online tutorials showed you exactly how to concatenate, but only if you already knew what the phrases were:
Tutorial: To concatenate x and y type x.=y
Lab Rat: I DON'T KNOW WHAT X AND Y ARE!!! In fact I'm looking for them because I don't know what they are. I want to find that out!;
Tutorial: *is no help at all*
Lab Rat: *Kicks computer, then hold up a large picture of a comma in front of the screen* Just find this, OK? See this picture, find something that looks like this and then give all the writing in front of it to me;
(the semicolon is computer for 'end of line'. I do not know why computers do this when almost every living person uses a full stop).
Computer: *Is not impressed*
I did actually get there in the end, to the surprise and delight of both myself and my supervisor (and probably the computer as well) I managed to get it to do sort of what I wanted. Unfortunately when we looked back over the raw data from our BLAST results we realised that there was a lot more information we wanted, so a lot more code has to be written. And here we hit another problem. The BLAST databases are truly amazing but just not particularly well organised. Some of them have the important information stored under /notes, while others have a separate field called /function. One item we saw even had the full protein function listed under /name. This means that to get all the information we need, we'll have to pull out each of these fields for every single protein, which will provide us with a lot of useless notes that we don't really need.
Lab Rat: Just give me the useful stuff, OK?;
Computer: Variable 'useful' not defined. Random computer squiggles, out of cheese error.
Lab Rat: *gives up on computers*;
Computer: *gives up on Lab Rat*
Some People Juggle Geese
I've spent most of today fighting with computer programmes, but during a brief lull (while pipetting out buffer L2 actually) I suddenly realised that there are an awful lot of odd things that you end up saying in a lab ranging from the slightly weird to the utterly confusing. So here's a little list. All of these have been said (or paraphrased) by me at some point in the last few weeks. And at the time they all made sense...
First, some that are just me being me:
"400 x 5 uh, yeah, that's 2"
"Oh shite, I've lost an order of magnitude"
"I'm going to pretend my lab coat is a trench coat"
"I'm actually beginning to doubt the existence of DNA."
"I can speak to computers!"
A couple of worrying ones:
"Ah. I didn't realise they were that expensive"
"Don't worry! I'm fine! The water just escaped."
"It seems that Agar at 50 degrees is not enough to remove fingerprints."
"Did you just hear the lid explode?"
And last, the vaguely baffling:
"My daisy is on the ice-box."
"Spot on seventy at a random twiddling of the twiddle."
"Shoogle it violently then go as fast as possible."
My favourite phrase is probably the "My daisy is on the ice-box" It has a slightly Monty Python-ish air to it.
First, some that are just me being me:
"400 x 5 uh, yeah, that's 2"
"Oh shite, I've lost an order of magnitude"
"I'm going to pretend my lab coat is a trench coat"
"I'm actually beginning to doubt the existence of DNA."
"I can speak to computers!"
A couple of worrying ones:
"Ah. I didn't realise they were that expensive"
"Don't worry! I'm fine! The water just escaped."
"It seems that Agar at 50 degrees is not enough to remove fingerprints."
"Did you just hear the lid explode?"
And last, the vaguely baffling:
"My daisy is on the ice-box."
"Spot on seventy at a random twiddling of the twiddle."
"Shoogle it violently then go as fast as possible."
My favourite phrase is probably the "My daisy is on the ice-box" It has a slightly Monty Python-ish air to it.
When you don't know who to trust...
To determine how much DNA is present in our DNA extractions, we use a neat little machine called a Nanodrop. This tells us how much DNA is present by giving us the absorption spectrums at A260 and A280 Abs. As DNA absorbs ultraviolet, this tells us what concentration of DNA we have.
It also tells us how contaminated our sample is. The ratio of the 260/280 should ideally be between 1.8 and 2. Any lower and there is significant protein contamination, and higher and there's probably lots of salt or something in there.
Or so we thought. And so quite a few of the references and pages seemed to suggest. Until, of course, we got to the Wikipedia article. This tells us, in no uncertain terms, that it takes a relatively large amount of protein contamination to significantly affect the 260:280 ratio, and even provides a little table to show that. It provides a citation link as well, which I can't get to. I've tried Google and Pubmed until I went round in circles, but no one wants to give me that paper. At least not for free :(
So who do we trust? The wikipedia article has a solid-looking table, whereas in most of the other things we read it was just a throwaway line. On the other hand "most of the other things we've read" includes the instruction manual for the machine, which should know what it's talking about. And I have yet to read the wikipedia citation.
On reflection, I think I will disbelieve Wikipedia this time. At any rate, it hardly matters because we still don't actually have any DNA.
Ah well. At least I'm getting paid for it =D
It also tells us how contaminated our sample is. The ratio of the 260/280 should ideally be between 1.8 and 2. Any lower and there is significant protein contamination, and higher and there's probably lots of salt or something in there.
Or so we thought. And so quite a few of the references and pages seemed to suggest. Until, of course, we got to the Wikipedia article. This tells us, in no uncertain terms, that it takes a relatively large amount of protein contamination to significantly affect the 260:280 ratio, and even provides a little table to show that. It provides a citation link as well, which I can't get to. I've tried Google and Pubmed until I went round in circles, but no one wants to give me that paper. At least not for free :(
So who do we trust? The wikipedia article has a solid-looking table, whereas in most of the other things we read it was just a throwaway line. On the other hand "most of the other things we've read" includes the instruction manual for the machine, which should know what it's talking about. And I have yet to read the wikipedia citation.
On reflection, I think I will disbelieve Wikipedia this time. At any rate, it hardly matters because we still don't actually have any DNA.
Ah well. At least I'm getting paid for it =D
Vaguely Spiritual
I was walking to the lab this morning, when I was stopped by a vaguely hippy-like woman handing out pamphlets. I always feel a bit sorry for people handing out pamphlets as they mostly get ignored, so I took one and decided to take a look at it. It was a religious one 'Does God care about suffering and if so why does it exist' or something like that.
My religious views can best be currently discribed as 'Single and Searching' so I decided to have a look through it. Before I go on to disect the thing though, I will make a quick disclaimer that unless your religious beliefs involve deliberately hurting people then I will respect them. I have no issue of any peoples of any religion, especially not christianity which I rather like because the Christian God actually had a go at being human for a while, which seems to me a sensible and charitable thing to do.
Religion is a touchy subject, but here I go anyway:
The Pamphlet
The first issue adressed was, unsurprisingly, does God exist. Unfortunately rather than going for the reasons I would have chosen (gap between the Mind and the body, the brain in a vat hypothesis, no fixed truth etc) they went for the Arguement from Design. Namely, everything is amazing, the world is so perfect therefore it must be designed. They even used Paley's wristwatch arguement, they went as far as to use the damn eye as well. This made me angry (actually it made me dissolve into giggles, but I should have been angry). Firstly because the whole eye arguement was debunked way back by Darwin, and secondly because if the eye is (as they claim) "Designed so that no camera could have done better" why are so many people wearing glasses? Was there a shortage of perfect eyes? Do only some people get them?
It had a nice phrase about the bible as well "No book has such credentials for historical accuracy". I'm still thinking about that and it baffles me no matter how I look at it.
My favourite bit though was the paragraph about the human 'unique blood system'. I can only presume they mean 'unique' in it's traditional sense of 'possessed by all vertebrates.' The quick science-babble about the blood system was, though, correct. Simplified but correct, which I don't mind at all. They just seemed to draw the oddest conclusions about it.
After all the waffle about the design of the earth, I was expecting it to go downhill from there. Surprisingly, it actually went uphill, dragging itself out of the shady science and giving a reasonable bit about free will. No doubt it would have annoyed the philosophers as much as the bad science annoyed me, but as a lay-person I didn't spot too many blinding errors.
Interestingly the picture of Adam and Eve in Eden had her handing over what was obviously and distinctly a pear. Not an apple. I've heard boths figs and bananas postulated as the actual fruit (my vote is with figs, personally) but never pears before.
After the free will part though, it seemed to take a bit of a fall again. Apparently, we are in the Last Days before the fall. This is because of the Wars, earthquakes and diseases that have been recently appearing in great quantities. It doesn't seem to have occured to the author that Wars, Earthquakes and diseases have always been happening in great quantities pretty much forever in human history. Seriously though, in English history (which I know most about) it's very hard to find a period of more than about 30 years without a war, disease, or natural disaster happening somewhere. Think of the plague, which wiped out almost 1/3 of the population.
It then sort of leaves the rails a bit and heads into lala land. Alright, probably not entirely fair, but I was a bit fed up with it at this point (the wristwatch arguement was still annoying me). The last section was talking happily about after judgement day, for a quick summary read through the last section of 'The Last Battle' by C.S Lewis, because what they were saying was pretty much word for word from the bit where Narnia is destroyed. All the good people will go through into this amazing 'new world', the sick will be better, the dead will come back to life, and presumably the space will be infinate because there are going to be an awful lot of people floating around. Oddly enough it never mentions what happens to be bad people, presumably we get to stay on whats left of the earth which, given the population will have gone down considerably, probably won't be too bad an option. We'll still get sick and die though.
They also mentioned who would be chosen as 'good' although they were remarkably cagey about it. I thought I was in the clear at first because all it was talking about was 'brotherly love' stuff and I'm quite a nice person. However at the end it sneaked in a clause about being and living 'under gods rule.'
Which, as I eat shellfish, means I'm stuffed. :(
My religious views can best be currently discribed as 'Single and Searching' so I decided to have a look through it. Before I go on to disect the thing though, I will make a quick disclaimer that unless your religious beliefs involve deliberately hurting people then I will respect them. I have no issue of any peoples of any religion, especially not christianity which I rather like because the Christian God actually had a go at being human for a while, which seems to me a sensible and charitable thing to do.
Religion is a touchy subject, but here I go anyway:
The Pamphlet
The first issue adressed was, unsurprisingly, does God exist. Unfortunately rather than going for the reasons I would have chosen (gap between the Mind and the body, the brain in a vat hypothesis, no fixed truth etc) they went for the Arguement from Design. Namely, everything is amazing, the world is so perfect therefore it must be designed. They even used Paley's wristwatch arguement, they went as far as to use the damn eye as well. This made me angry (actually it made me dissolve into giggles, but I should have been angry). Firstly because the whole eye arguement was debunked way back by Darwin, and secondly because if the eye is (as they claim) "Designed so that no camera could have done better" why are so many people wearing glasses? Was there a shortage of perfect eyes? Do only some people get them?
It had a nice phrase about the bible as well "No book has such credentials for historical accuracy". I'm still thinking about that and it baffles me no matter how I look at it.
My favourite bit though was the paragraph about the human 'unique blood system'. I can only presume they mean 'unique' in it's traditional sense of 'possessed by all vertebrates.' The quick science-babble about the blood system was, though, correct. Simplified but correct, which I don't mind at all. They just seemed to draw the oddest conclusions about it.
After all the waffle about the design of the earth, I was expecting it to go downhill from there. Surprisingly, it actually went uphill, dragging itself out of the shady science and giving a reasonable bit about free will. No doubt it would have annoyed the philosophers as much as the bad science annoyed me, but as a lay-person I didn't spot too many blinding errors.
Interestingly the picture of Adam and Eve in Eden had her handing over what was obviously and distinctly a pear. Not an apple. I've heard boths figs and bananas postulated as the actual fruit (my vote is with figs, personally) but never pears before.
After the free will part though, it seemed to take a bit of a fall again. Apparently, we are in the Last Days before the fall. This is because of the Wars, earthquakes and diseases that have been recently appearing in great quantities. It doesn't seem to have occured to the author that Wars, Earthquakes and diseases have always been happening in great quantities pretty much forever in human history. Seriously though, in English history (which I know most about) it's very hard to find a period of more than about 30 years without a war, disease, or natural disaster happening somewhere. Think of the plague, which wiped out almost 1/3 of the population.
It then sort of leaves the rails a bit and heads into lala land. Alright, probably not entirely fair, but I was a bit fed up with it at this point (the wristwatch arguement was still annoying me). The last section was talking happily about after judgement day, for a quick summary read through the last section of 'The Last Battle' by C.S Lewis, because what they were saying was pretty much word for word from the bit where Narnia is destroyed. All the good people will go through into this amazing 'new world', the sick will be better, the dead will come back to life, and presumably the space will be infinate because there are going to be an awful lot of people floating around. Oddly enough it never mentions what happens to be bad people, presumably we get to stay on whats left of the earth which, given the population will have gone down considerably, probably won't be too bad an option. We'll still get sick and die though.
They also mentioned who would be chosen as 'good' although they were remarkably cagey about it. I thought I was in the clear at first because all it was talking about was 'brotherly love' stuff and I'm quite a nice person. However at the end it sneaked in a clause about being and living 'under gods rule.'
Which, as I eat shellfish, means I'm stuffed. :(
PayDay
I have just received my first paycheck. This is quite a big moment for me; for the first time ever someone has given me money in order to carry out science, thus making me (in my eyes at least) a Proper Scientist.
I chose to celebrate this great moment in my lifetime by spinning around very fast on my computer chair making squeaky noises. I still can't quite believe that someone has handed me a substantial amount of money for the most enjoyable six weeks of my holiday.
And the best thing is that due to an accumulation of reasons within the lab (and my long summer holiday) I have been allowed to stay a Lab Rat in September as well.
eeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeee!
As it would probably be a good idea to put something at least vaguely scientific in this post I tried to look up how autoclave tape is made. Autoclave tape looks exactly like masking tape except that it has fine white lines across it. It is also apparently slightly more sticky. You attach it to anything that you are about to put in the autoclave (which heats things up to very high temperatures in order to sterilize them) and as they are autoclaved the white lines go black. This allows you to tell instantly whether or not something has been autoclaved (and is also handy for holding stuff together).
Wikipedia says this:
"Autoclave tape is an adhesive tape used in autoclaving to indicate whether the correct temperature has been reached for the elimination of all living organisms (typically 121 degrees Celsius).[1]
I chose to celebrate this great moment in my lifetime by spinning around very fast on my computer chair making squeaky noises. I still can't quite believe that someone has handed me a substantial amount of money for the most enjoyable six weeks of my holiday.
And the best thing is that due to an accumulation of reasons within the lab (and my long summer holiday) I have been allowed to stay a Lab Rat in September as well.
eeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeee!
As it would probably be a good idea to put something at least vaguely scientific in this post I tried to look up how autoclave tape is made. Autoclave tape looks exactly like masking tape except that it has fine white lines across it. It is also apparently slightly more sticky. You attach it to anything that you are about to put in the autoclave (which heats things up to very high temperatures in order to sterilize them) and as they are autoclaved the white lines go black. This allows you to tell instantly whether or not something has been autoclaved (and is also handy for holding stuff together).
Wikipedia says this:
"Autoclave tape is an adhesive tape used in autoclaving to indicate whether the correct temperature has been reached for the elimination of all living organisms (typically 121 degrees Celsius).[1]
Small strips of the tape are applied to the items before they are placed into the autoclave. The tape is similar to masking tape but slightly more adhesive, to allow it to adhere under the hot, moist conditions of the autoclave. The tape typically has diagonal markings containing an ink which changes colour (usually beige to black) upon heating. One such ink contains 30.1% lead thiosulfate, 0.6% magnesium carbonate, 20.1% neocryl B8141, 30.1% ethanol, 22.7% ethyl acetate and 49% ink solids. Unfortunately these percentages add up to more than 100%, so this data is completely bogus. "
Yes. Very helpful. The manufacturers aren't particularly keen on giving the secret away either. This maybe for complicated legal reasons or it may just be that it hasn't occurred to them that anybody would be interested in knowing what autoclave tape is made of.
K
There's a slight problem at the lab at the moment. We aren't getting any DNA. The lovely little DNA extraction protocol is not actually extracting huge amounts of DNA for us, if fact it seems to be producing pretty much no DNA for us.
We've checked through the troubleshooting, and in various technique manuals, and found a couple of modifying features to add to our protocol, which will hopefully make it more productive. Most of them are fairly basic (longer incubation's etc.) but the one that caught my eye was the addition of proteinase K at the denaturing step. When you add SDS (which breaks down all the phage proteins to supposedly release the DNA) you also add this mystery stuff called proteinase K.
Not wanting something so scientific to be shooting over my Magical Event Horizon, I went to take a look at what it was. A protease is a substance that breaks down proteins, and it turns out that this is exactly what proteinase K does, it is far more active when surrounded by SDS as well, so hopefully the combination of the two will pretty much shred all the phage proteins releasing all the DNA (as I said, phages are nothing but DNA and protein).
One interesting thing I did find though is that proteinase K was extracted from a fungus (called Tritirachium album Limber). It's fairly deadly as well, it can pull apart all sorts of different proteins. It makes sense for a fungi to possess a powerful proteinase like this as fungi are saprotrophic feeders; they release the enzymes to digest their food outside of their actual bodies and then just absorb all the nutrients left behind.
I thought that the name 'proteinase K' might turn out to have a fairly interesting backstory (why K?) but it is, in fact, spectacularly boring. It was named Protienase K due to its keratin hydrolyzing activity which, seeing as it hydrolyses (breaks down) a large number of proteins seems fairly arbitrary. Biologists have very little imagination sometimes.
If you have a desperate longing to get hold of some proteinase K you can buy it here.
It is fairly expensive though; £39.00 for 2ml.
We've checked through the troubleshooting, and in various technique manuals, and found a couple of modifying features to add to our protocol, which will hopefully make it more productive. Most of them are fairly basic (longer incubation's etc.) but the one that caught my eye was the addition of proteinase K at the denaturing step. When you add SDS (which breaks down all the phage proteins to supposedly release the DNA) you also add this mystery stuff called proteinase K.
Not wanting something so scientific to be shooting over my Magical Event Horizon, I went to take a look at what it was. A protease is a substance that breaks down proteins, and it turns out that this is exactly what proteinase K does, it is far more active when surrounded by SDS as well, so hopefully the combination of the two will pretty much shred all the phage proteins releasing all the DNA (as I said, phages are nothing but DNA and protein).
One interesting thing I did find though is that proteinase K was extracted from a fungus (called Tritirachium album Limber). It's fairly deadly as well, it can pull apart all sorts of different proteins. It makes sense for a fungi to possess a powerful proteinase like this as fungi are saprotrophic feeders; they release the enzymes to digest their food outside of their actual bodies and then just absorb all the nutrients left behind.
I thought that the name 'proteinase K' might turn out to have a fairly interesting backstory (why K?) but it is, in fact, spectacularly boring. It was named Protienase K due to its keratin hydrolyzing activity which, seeing as it hydrolyses (breaks down) a large number of proteins seems fairly arbitrary. Biologists have very little imagination sometimes.
If you have a desperate longing to get hold of some proteinase K you can buy it here.
It is fairly expensive though; £39.00 for 2ml.
I'm in less of a sciency mood...
...because I've just started watching Firefly. wow. such amazing characters. Such amazing character interactions. Such an awesome mix of sci-fi and western without clogging it with unnecessary romance. Seriously though, is there any other sci-fi movie where the pilot is in a loving and stable marriage? Or where explosions in space happen to the accompaniment of awesome guitar music rather than actual scientifically-incorrect explosions?
On the lab front, we are busy troubleshooting. The DNA extraction technique is extracting tiny amounts of DNA and the task today is to find out why. Which means going through the whole process all over again (it takes about six hours) extracting aliquots every time we so much as pick up an eppindorf and try to find out where exactly our DNA is going.
...
They never have problems like this on Serenity.
On the lab front, we are busy troubleshooting. The DNA extraction technique is extracting tiny amounts of DNA and the task today is to find out why. Which means going through the whole process all over again (it takes about six hours) extracting aliquots every time we so much as pick up an eppindorf and try to find out where exactly our DNA is going.
...
They never have problems like this on Serenity.
Agarose gel
Today's task is running gels, to see if our DNA extraction technique managed to extract DNA. Halfway through measuring out the agarose for the gel base I suddenly realised that I had very little idea of how these gels actually worked. Lab Rat job of the day therefore, was to find out more about them (hopefully in order to prevent further embarrassment of the NrdG kind).
Agarose gels
Agarose gel electrophoresis is a basic method used to separate RNA and DNA of different sizes. The gel itself is made up of an agarose matrix (formed from agarose, ethidium bromide and TAE; Tris-acetate EDTA. More on those later) with little wells at the top to add the DNA. As DNA is negatively charged, you can run an electric charge across the gel causing all the DNA to migrate down it.
The agarose and TAE make up a matrix. The TAE acts as a buffer, keeping the agarose at a constant pH. The agarose makes the gel more difficult for the DNA to run through; which allows the DNA to separate. Small fragments can get through relatively easily while larger fragments take a lot longer.
Ethidium bromide is used to visualise the gels. It fluoresces under UV light when intercalated into DNA, allowing you to visualise any band containing ~20ng of DNA under a UV lamp. The problem with ethidium bromide is that it intercalates with any DNA, including the DNA on (say) your skin. This makes it a powerful carcinogen, so it's a good idea to wear gloves and coats while handling it.
As well as length, the distance moved by the DNA is also affected by its conformation. Linear fragments sort nice smart bands (ideally) while circular DNA just forms a sort of elongated blob. Because of this, circular DNA fragments (i.e from plasmids or viruses) are usually cut up using restriction enzymes. As we're just seeing if we've got any DNA at the moment, we haven't bothered cutting it up into pieces, we're just running the circular DNA from our viruses.
All fun stuff :)
Agarose gels
Agarose gel electrophoresis is a basic method used to separate RNA and DNA of different sizes. The gel itself is made up of an agarose matrix (formed from agarose, ethidium bromide and TAE; Tris-acetate EDTA. More on those later) with little wells at the top to add the DNA. As DNA is negatively charged, you can run an electric charge across the gel causing all the DNA to migrate down it.
The agarose and TAE make up a matrix. The TAE acts as a buffer, keeping the agarose at a constant pH. The agarose makes the gel more difficult for the DNA to run through; which allows the DNA to separate. Small fragments can get through relatively easily while larger fragments take a lot longer.
Ethidium bromide is used to visualise the gels. It fluoresces under UV light when intercalated into DNA, allowing you to visualise any band containing ~20ng of DNA under a UV lamp. The problem with ethidium bromide is that it intercalates with any DNA, including the DNA on (say) your skin. This makes it a powerful carcinogen, so it's a good idea to wear gloves and coats while handling it.
As well as length, the distance moved by the DNA is also affected by its conformation. Linear fragments sort nice smart bands (ideally) while circular DNA just forms a sort of elongated blob. Because of this, circular DNA fragments (i.e from plasmids or viruses) are usually cut up using restriction enzymes. As we're just seeing if we've got any DNA at the moment, we haven't bothered cutting it up into pieces, we're just running the circular DNA from our viruses.
All fun stuff :)
Magical Event Horizon
One concept I vaguelly mentioned yesterday, while going through the DNA extraction protocol, was that of the Magical Event Horizon. I love this concept (which I first read in a Terry Pratchett book but anyway) which works on the Arthur C. Clarke quote that "sufficiently advanced technology is indistinguishable from magic".
The idea is, therefore, that everyone understands things up to a point. That point is the Magical Event Horizon (or MEH). For me, for example, bacteriophages and calculus are well within the MEH, relativity is sort of dancing along the line and the inner workings of computers are far, far over my Magical Event Horizon.
It's a neat little concept. Anything that you personally sort of feel to yourself works by 'magic' (like digital cameras. I have no idea how those work) goes over the MEH, everything you can explain stays within it.
The idea is, therefore, that everyone understands things up to a point. That point is the Magical Event Horizon (or MEH). For me, for example, bacteriophages and calculus are well within the MEH, relativity is sort of dancing along the line and the inner workings of computers are far, far over my Magical Event Horizon.
It's a neat little concept. Anything that you personally sort of feel to yourself works by 'magic' (like digital cameras. I have no idea how those work) goes over the MEH, everything you can explain stays within it.
DNA Extraction
Todays task is extracting DNA from our T4 phages that we've spent the last two weeks growing and nurturing and caring for. To make this easier, we're using a ready-made Kit called QIAGEN (which I keep trying to pronounce qui-gon :D ). QIAGEN basically provides all the material and gives an idiot guide as to how to use it, which means that Lab Rat's task of the day involves going through said protocol and working out what is actually happening in each step.
First though, another quick notice from the Department of the Very Obvious:
---
DNA Extraction
1.Grow the phages on agarose gel. Agar gel is a gelatenous substance from seaweed that contains both agarose and agaropectin. Agaropectin contains lots of acidic sidegroups (containings sulfur and various other things like pyruvate). Agar is usually used for gels as it's cheaper, but for DNA extraction you can't risk any of the acidic agaropectin loitering around as it stops the extraction enzymes from working. Instead you use just pure agarose.
2.Isolate phages. This is done by peeling off the top layer of agarose and using a centrifuge to spin it down to the bottom, leaving the phages in the supernatant (the liquid left behind after centrifuging). Push the supernatant through a couple of filters to remove anything else (bacteria mostly) and you're left with a pure phage solution.
3.Now the DNA extraction can start. The first instruction in the kit is 'add buffer L1'. Buffer L1 is a clever mixture of various different enzymes and buffer solutions which breaks down any bacterial RNA or DNA that might be left in your solution.
4.Next buffer L2 is added. This precipitates out the phage particles; essentially it clumps all the bacteriophage's together making them easier to extract.
5.Centrifuge to collect the phage. The centrifuge is the big fast-spinning machine that pellets all the phages down into a neat little, well, pellet. Very useful machine, and it would be even more useful if ours worked properly :(
6.Resuspend the phage in buffer L3. I suspect this is just a growth medium, to turn the phage pellet into a phage suspension.
7.Add buffer L4. Buffer L4 contains the well known SDS (which comes up very often in various extractions). SDS is a detergent which essentially breaks all the proteins down. As viruses pretty much just consist of proteins and DNA this means that the only whole thing left in the test-tube at this point is the DNA. Still quite a way to go though, so here's a diagram of a centrifuge: 8. Add buffer L5, mix and centrifuge. It does not actually say, but i suspect L5 is precipitating the proteins that have just been cut up. The centrifuge will then pellet the proteins down to separate them from our DNA.
8. Add buffer L5, mix and centrifuge. It does not actually say, but i suspect L5 is precipitating the proteins that have just been cut up. The centrifuge will then pellet the proteins down to separate them from our DNA.
9. Equilibrate a QIAGEN-tip 20 by adding buffer QBT. This is where we hit the Magic Event Horizon (MEH) at top speed. I have no idea what is in the buffer or exactly how a QIAGEN-tip works, which is probably a good thing for copyright in general. You can get to QIAGEN to find out more about it by going here: QIAGEN
10. Allow the residue from step 8 to flow through the QIAGEN tip. One thing I do know about the tips is that they contain resin. The resin traps the DNA on it allowing the rest of the phage to wash through (essentially this will just be any liquid medium, as all the proteins have technically been removed by the centrifuge in step 8)
11. Wash the QIAGEN tip with buffer QC. This washes the DNA (which is trapped on the filter) removing any last impurities.
12. Collect DNA with buffer QC into a clean tube. The QC in some way (Magical Event Horizon fast approaching) allows the DNA to flow through the filter and into the new tube. Finally the DNA! We now (should) have a solution containing nothing but phage DNA. Just got to collect it.
13. Precipitate the DNA by adding isopropanol. Isopropanol is another old favourite, it just clumps DNA, making very very hard-to-see pellets. You've probably guessed by now but the next stage is: centrifuging, to collect the pellet.
14. Wash pellet with ethanol. Ethanol removes any residual salt as there will be some magnesium and various others in the DNA (DNA has an overall negative charge due to the phosphate backbone which collects positively charged salts). Washing involves adding ethanol, shaking very gently and then (surprise surprise) centrifuging to pellet the DNA again and remove all the ethanol.
15. Allow DNA to dry and redissolve in buffer. Buffer just keeps the DNA happy and stops it disintegrating.
And that's it! We now have a little glass bottle containing a solution of pure DNA.
Science really is just like cookery =D

First though, another quick notice from the Department of the Very Obvious:
- Do not try to do DNA extractions when hung-over
---
DNA Extraction
1.Grow the phages on agarose gel. Agar gel is a gelatenous substance from seaweed that contains both agarose and agaropectin. Agaropectin contains lots of acidic sidegroups (containings sulfur and various other things like pyruvate). Agar is usually used for gels as it's cheaper, but for DNA extraction you can't risk any of the acidic agaropectin loitering around as it stops the extraction enzymes from working. Instead you use just pure agarose.
2.Isolate phages. This is done by peeling off the top layer of agarose and using a centrifuge to spin it down to the bottom, leaving the phages in the supernatant (the liquid left behind after centrifuging). Push the supernatant through a couple of filters to remove anything else (bacteria mostly) and you're left with a pure phage solution.
3.Now the DNA extraction can start. The first instruction in the kit is 'add buffer L1'. Buffer L1 is a clever mixture of various different enzymes and buffer solutions which breaks down any bacterial RNA or DNA that might be left in your solution.
4.Next buffer L2 is added. This precipitates out the phage particles; essentially it clumps all the bacteriophage's together making them easier to extract.
5.Centrifuge to collect the phage. The centrifuge is the big fast-spinning machine that pellets all the phages down into a neat little, well, pellet. Very useful machine, and it would be even more useful if ours worked properly :(
6.Resuspend the phage in buffer L3. I suspect this is just a growth medium, to turn the phage pellet into a phage suspension.
7.Add buffer L4. Buffer L4 contains the well known SDS (which comes up very often in various extractions). SDS is a detergent which essentially breaks all the proteins down. As viruses pretty much just consist of proteins and DNA this means that the only whole thing left in the test-tube at this point is the DNA. Still quite a way to go though, so here's a diagram of a centrifuge:
 8. Add buffer L5, mix and centrifuge. It does not actually say, but i suspect L5 is precipitating the proteins that have just been cut up. The centrifuge will then pellet the proteins down to separate them from our DNA.
8. Add buffer L5, mix and centrifuge. It does not actually say, but i suspect L5 is precipitating the proteins that have just been cut up. The centrifuge will then pellet the proteins down to separate them from our DNA.9. Equilibrate a QIAGEN-tip 20 by adding buffer QBT. This is where we hit the Magic Event Horizon (MEH) at top speed. I have no idea what is in the buffer or exactly how a QIAGEN-tip works, which is probably a good thing for copyright in general. You can get to QIAGEN to find out more about it by going here: QIAGEN
10. Allow the residue from step 8 to flow through the QIAGEN tip. One thing I do know about the tips is that they contain resin. The resin traps the DNA on it allowing the rest of the phage to wash through (essentially this will just be any liquid medium, as all the proteins have technically been removed by the centrifuge in step 8)
11. Wash the QIAGEN tip with buffer QC. This washes the DNA (which is trapped on the filter) removing any last impurities.
12. Collect DNA with buffer QC into a clean tube. The QC in some way (Magical Event Horizon fast approaching) allows the DNA to flow through the filter and into the new tube. Finally the DNA! We now (should) have a solution containing nothing but phage DNA. Just got to collect it.
13. Precipitate the DNA by adding isopropanol. Isopropanol is another old favourite, it just clumps DNA, making very very hard-to-see pellets. You've probably guessed by now but the next stage is: centrifuging, to collect the pellet.
14. Wash pellet with ethanol. Ethanol removes any residual salt as there will be some magnesium and various others in the DNA (DNA has an overall negative charge due to the phosphate backbone which collects positively charged salts). Washing involves adding ethanol, shaking very gently and then (surprise surprise) centrifuging to pellet the DNA again and remove all the ethanol.
15. Allow DNA to dry and redissolve in buffer. Buffer just keeps the DNA happy and stops it disintegrating.
And that's it! We now have a little glass bottle containing a solution of pure DNA.
Science really is just like cookery =D
Things I've learnt today...
If you're writing a report, don't write the discussion in five minutes straight. And don't print immediately after writing the discussion. And don't hand the report in without checking you've written everything you want to write.
uuurgh...
I had lots of interesting things to put in that discussion as well. It was going to be a good discussion.
Other things I've learnt:
:(
Good news though: We have phage! They grew, with no contamination, which means that tommorrow we get to slice them open and take their DNA out. YAY!!
uuurgh...
I had lots of interesting things to put in that discussion as well. It was going to be a good discussion.
Other things I've learnt:
- Don't waste time looking at random sites when you should be writing reports
- Don't do this so often that you end up writing the report the morning it's due
- Don't miss breakfast to write a report
- The pope is a catholic
- Bears really do shit in the woods.
:(
Good news though: We have phage! They grew, with no contamination, which means that tommorrow we get to slice them open and take their DNA out. YAY!!
Contamination!
 I came in this morning, all bright and ready for phage propagation (harvesting phages from plates is actually quite fun) and what did we find? All the plates from yesterday were bad. To make a phage plaque you plate out a smooth 'lawn' of bacteria onto an agar plate and then pour phage over it. Where one phage lands it kills and multiplies until it forms a little clear spot or 'plaque' on the plate. (you can't see the phage plaques very well in the picture, but you can see the bacterial lawn)
I came in this morning, all bright and ready for phage propagation (harvesting phages from plates is actually quite fun) and what did we find? All the plates from yesterday were bad. To make a phage plaque you plate out a smooth 'lawn' of bacteria onto an agar plate and then pour phage over it. Where one phage lands it kills and multiplies until it forms a little clear spot or 'plaque' on the plate. (you can't see the phage plaques very well in the picture, but you can see the bacterial lawn)This morning, all our plates had little fuzzy marks on them. Which is fine, could be phages except...so did the control plate. The control plate had no phage on it at all, it was meant to have just the bacteria but itstead it too had little fuzzy marks.
*sigh*
Without good controls you can't trust your results. At first we thought it was just badly grown host until my supervisor noticed that the tryptone we'd been working with (tryptone broth is one medium that you can grow the bacteria in) was cloudy. Not good. If a substance that is usually clear starts looking cloudy then it means it's got bacteria in it.
Our plates were all contaminated. The fuzzy marks were phage, bad host and/or another bacterial contaminant. Basically we have to do all of yesterdays work all over again today and we need a lot more media (agar for plates, tryptone for bacteria, etc).
Guess who gets to make up all the new media?
I do love working in labs. But it can sometimes be the more frustrating thing on earth. And media pouring is just mindnumbingly boring and faintly awkward. Which is why, of course, the lab rat gets to do it.
How to look like an idiot...
A fairly embarrassing thing happened yesterday afternoon. My supervisor's PI (the one who owns the lab and is in charge of all the research) came by to see how we were doing and he asked me about my bioinformatics project. My bioinformatics project involves looking at bits DNA from our bacteriophage and trying to work out what it does, using all sorts of clever internet search programs (mostly BLAST actually).
The conversation went something like this:
PI: So, hows the work going?
Lab Rat: Fine. I'm pretty sure about the first sequence, it's got a fairly obvious NrdG domain, and it's really conserved with other NrdG domains. I reckon it's a NrdG.
PI: Great. So what does an NrdG domain do? Why have we got one in the phage?
Lab Rat: *frantically scans notes*. Uh, an NrdG domain, uh, ribonucleotide reductase, uh, class III anaerobic ribonucleotide reductase. Um. assembles deoxyribonucleotides.
PI: ...
Lab Rat: I'll look it up.
So my first task this morning was heading over to PubMed. For those not in the know, PubMed is the worlds most wonderful search engine, which scans all the science reviews and papers and can usually find the one you want. For those who are interested, here is a brief overview of what NrdG domains are:
The conversation went something like this:
PI: So, hows the work going?
Lab Rat: Fine. I'm pretty sure about the first sequence, it's got a fairly obvious NrdG domain, and it's really conserved with other NrdG domains. I reckon it's a NrdG.
PI: Great. So what does an NrdG domain do? Why have we got one in the phage?
Lab Rat: *frantically scans notes*. Uh, an NrdG domain, uh, ribonucleotide reductase, uh, class III anaerobic ribonucleotide reductase. Um. assembles deoxyribonucleotides.
PI: ...
Lab Rat: I'll look it up.
So my first task this morning was heading over to PubMed. For those not in the know, PubMed is the worlds most wonderful search engine, which scans all the science reviews and papers and can usually find the one you want. For those who are interested, here is a brief overview of what NrdG domains are:
NrdG
NrdG is a domain in a protein, which basically means that it is an area of the protein with a recognisable structure. Search engines such as InterProScan are designed to look for these domains in proteins and show you where they are. You can then (if you do your research properly) go to PubMed and find out what the domain does.
One of the most important tasks in a cell is the replication of cellular DNA. DNA is built up from small subunits, or monomers, known as deoxyribonucleotides. In order to replicate the DNA the cell must have enough of these monomers to make a new strand of DNA. However most cells do not replicate all the time, at different times in their life cycles they will need different concentrations of deoxyribonucleotides inside the cell.
Enter ribonucleotide reductase: This controls the concentration of DNA monomers within the cell. It works by using oxygen to generate a tyrosyl radical (basically just a very reactive molecule, radicals are very reactive and have been shown to play an important role in catalysis). The tyrosyl radical then catalysis the reaction, producing new DNA monomers. Control of ribonucleotide reductase therefore, controls the amount of DNA monomers.
The problem with this is that some bacteria don't use oxygen. These are known as anaerobic bacteria. Without oxygen, they cannot generate the tyrosyl radical, which is vital for the reaction to occur. So how do they manage it? This (finally) is where my NrdG domain comes in. The NrdG domain contains a class III reductase, which is an anaerobic reductase. Instead of using oxygen to create a tyrosyl radical, it uses a different system to produce a glycyl radical. Still a radical (so it still has high reactive power) but made from a different molecule (glycerol rather than tyrosine).
The golden question is of course, what is this doing in my phage? Bacteriophages don't make DNA, they invade bacteria and use that bacteria to make DNA for them. Also, my bacteria is a lytic phage which means that it's only in the bacteria for a short amount of time before killing it. (unlike lysogenic phages, which hang around in the bacteria for long lengths of time. Lysogenic phages often carry bits of DNA which their bacterial hosts will find useful).
For a phage to carry a ribonucleotide reductase is not, however, such a bad idea. The host for my phage is a bacteria which can survive in both aerobic (with oxygen) and anaerobic (without oxygen) conditions. It is plausible, therefore, that the phage carries with it a gene that helps the bacteria to make DNA, especially in anaerobic conditions, when most cellular processes tend to be slower anyway. That way the phage can get its DNA made up and packaged as quickly as possible and then break out of the bacteria.
Feel free to ask any questions in the comments :)
One of the most important tasks in a cell is the replication of cellular DNA. DNA is built up from small subunits, or monomers, known as deoxyribonucleotides. In order to replicate the DNA the cell must have enough of these monomers to make a new strand of DNA. However most cells do not replicate all the time, at different times in their life cycles they will need different concentrations of deoxyribonucleotides inside the cell.
Enter ribonucleotide reductase: This controls the concentration of DNA monomers within the cell. It works by using oxygen to generate a tyrosyl radical (basically just a very reactive molecule, radicals are very reactive and have been shown to play an important role in catalysis). The tyrosyl radical then catalysis the reaction, producing new DNA monomers. Control of ribonucleotide reductase therefore, controls the amount of DNA monomers.
The problem with this is that some bacteria don't use oxygen. These are known as anaerobic bacteria. Without oxygen, they cannot generate the tyrosyl radical, which is vital for the reaction to occur. So how do they manage it? This (finally) is where my NrdG domain comes in. The NrdG domain contains a class III reductase, which is an anaerobic reductase. Instead of using oxygen to create a tyrosyl radical, it uses a different system to produce a glycyl radical. Still a radical (so it still has high reactive power) but made from a different molecule (glycerol rather than tyrosine).
The golden question is of course, what is this doing in my phage? Bacteriophages don't make DNA, they invade bacteria and use that bacteria to make DNA for them. Also, my bacteria is a lytic phage which means that it's only in the bacteria for a short amount of time before killing it. (unlike lysogenic phages, which hang around in the bacteria for long lengths of time. Lysogenic phages often carry bits of DNA which their bacterial hosts will find useful).
For a phage to carry a ribonucleotide reductase is not, however, such a bad idea. The host for my phage is a bacteria which can survive in both aerobic (with oxygen) and anaerobic (without oxygen) conditions. It is plausible, therefore, that the phage carries with it a gene that helps the bacteria to make DNA, especially in anaerobic conditions, when most cellular processes tend to be slower anyway. That way the phage can get its DNA made up and packaged as quickly as possible and then break out of the bacteria.
Feel free to ask any questions in the comments :)
Subscribe to:
Posts (Atom)